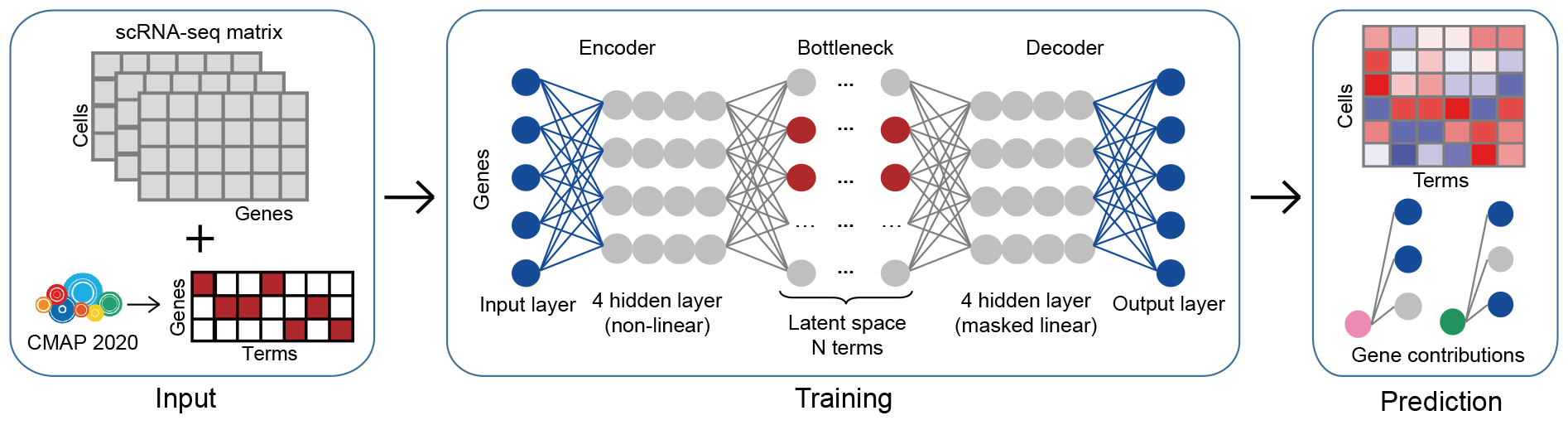
The Shennong framework consists of three main stages. In brief, we first integrated the scRNA-seq count matrix with preprocessed perturbation data to obtain the perturbation changes on each cell and transformed it into a binary matrix that represented the genetic change of one term (treatment) in each cell. The perturbation matrix was extracted from the high-confidence signatures of level 5 in CMap 2020 version, which consists of ~8 billion gene-expression profiles on the responses of ~240 human cell lines to over ~39,320 compounds across a range of concentrations, retaining features that were significantly differentially expressed in each treatment (Supplementary Fig. 9a).

Next, to explore and query the effects of perturbations on each cell, we used scRNA-seq gene expression profiles and condition labels for each cell to encode a set of terms, adopted variational autoencoder architecture to prune and enrich terms and decoder architecture to explain the genetic contributions of each term, with the latent space dimension equal to the number of terms. The model leverages a nonlinear encoder for flexibility and a linear decoder for interpretability, based on the publicly available model expiMap. Considering the large scale and potential redundancy of perturbation data, the attention-like mechanism was implemented in latent space to select only perturbation influence terms on each cell (see Materials and Methods).
Finally, based on the trained end-to-end model, we could predict the variation induced by term in the query data, as well as the measure affection of the genes in each term. The proposed framework takes advantage of large-scale perturbation datasets to explore and capture the gene expression variation induced by a diverse array of compounds at the single-cell level and quantify the contribution of affected genes in these variations.
Shennong source code (github): https://github.com/PeijingZhang/Shennong.
Perturbation data: perturbation.gmt, perturbation.gmt.h5ad (high-confidence signatures of CMap with already preproceessed)
Example data: adata_train.h5ad, adata_predict.h5ad, command_example.sh, example.ipynb
Script: preprocess.py, train.py, predict.py
To discover candidate drugs and their potential targets, we applied Shennong to our pan-cancer landscape and explored the response of the tumor cells to pharmacologic compounds.
Based on the latent scores and directions of terms in each cell, we identified the significantly differential terms in each cell type. The term was a collection of significant features extracted from perturbation data that reflect the effects of the compound treatment on cells.
To investigate mechanisms of drug action, we investigated the contribution of individual genes in each term, measuring the comparative importance of genes within each term, and helping to elucidate potential targets of corresponding compounds, associated signaling and metabolic pathways.